



The importance of temperature within the pharmaceutical industry
Regulations
The World Health Organisation (WHO) develops international norms and standards to regulate health products and to eliminate substandard medicines, but must pharmaceutical manufacturers comply to the WHO norms?
The International Council for Harmonisation for Technical Requirements for Pharmaceuticals for Human Use (ICH) brings together the regulatory authorities and the pharmaceutical companies and develop ICH guidelines that are applied by the regulatory authorities. A regulatory authority is a public or government agency responsible that creates rules and regulations for a specific area of application, each country will have their own regulatory authority and as of such, their own rules and regulations.
The best-known authorities are the US Food and Drug Administration (FDA) and the EudraLex – EU Legislation regulating who can manufacture, stock and distribute medicinal products. Medicinal products may be manufactured in one country but are imported into another: these products are subjected to the receiving country’s authority. So, it makes sense that all regulatory authorities are aligned.
However, as the official regulations leave room for interpretation, organisations have been created to ensure that all regulated companies understand and comply to the regulations:
- The International Society for Pharmaceutical Engineering (ISPE),
- The US Pharmacopeia (USP),
- The World Health Organisation (WHO).
The International Organisation for Standardization (ISO) is an independent, international organisation that develops international standards. Standards are voluntary and not mandatory, however, regulatory authorities many use standards to support technical regulations.
So, no, pharmaceutical manufacturers do not have to comply to the WHO norms and standards to deal with pharmaceutical products. The regulated company must comply with the regulations defined by the local regulatory authority and can then use the WHO norms and standards to help comply with the regulations.
Temperature defines product quality
When certain products are exposed to incorrect temperatures the product quality is no longer guaranteed. In the pharmaceutical, cosmeceutical and the food industry, this is even more important as consumers lives can be put at risk due to the temperature effects on the products/ingredients such as decomposition, diminished effects/taste and spoilage.
In the pharmaceutical industry regulations have been put in place to protect the patients and to deliver quality-assured, safe and effective products. The temperature range, along with other parameters, to ensure product quality is defined during the product risk assessment.
What are pharmaceutical products:
Drugs, medicines, vaccines active pharmaceutical ingredients (API), human samples, bio-pharmaceuticals, medical devices…
Applications
Here is an overview of the applications where temperature monitoring is crucial:
- Cryogenic monitoring.
- Ultra-low temperatures monitoring (including with dry ice).
- Fridge/freezer monitoring.
- Stability testing.
- Incubators monitoring.
- Storage monitoring.
- Warehouse monitoring.
- Distribution centre monitoring
- Cleanroom monitoring.
- Transport monitoring (trucks, airplanes, containers, trains…)
- Healthcare sales point monitoring (pharmacies, vets, dentists, hospitals…)
- Blood bank monitoring
- Tissue bank monitoring
- Biobank monitoring
- Calibration laboratory monitoring
Quality Guidelines
The GxP quality guidelines, developed by the regulatory authorities, are designed to cover the product quality over the complete life cycle of the product.

Initially during a new medicine development phase:

Once the R & D phase is completed and the pharmaceutical product is approved by the regulatory entity, production is scaled up and the medicines are available to patients

Good Laboratory Practice (GLP): Defines a set of rules and criteria for a quality system concerned with the organisational process and the conditions under which non-clinical health and environmental safety studies are planned, performed, monitored, recorded, reported and archived.
Good Distribution Practice (GDP): Describes the minimum standards that must be met to ensure that the quality and integrity of medicines is maintained throughout the supply chain. GDP applies to sourcing, storage and transportation of active pharmaceutical ingredients and other ingredients used in the production of medicines.
Good Manufacturing Practice (GMP): Describes the minimum standards that a medicines manufacturer must meet in their production processes. For the example of GMP, here are the guidelines from the FDA and EU:
- FDA 21 CFR Part 210: Current Good Manufacturing Practice in Manufacturing Process, packing, or Holding of Drugs.
- FDA 21 CFR Part 211: Current Good Manufacturing for Finished Pharmaceuticals.
- EudraLex – Volume 4 – Good Manufacturing Practice (GMP) Guidelines.
Complying to the GxP regulations
The GxP guidelines state that the area used must be qualified, temperature should be monitored, and monitoring devices must be calibrated to meet the guidelines.
Qualified: The qualification of the area is the process of proving that the area meets the requirements for its intended purpose.
Monitored: The critical control parameters (temperature amongst others) are measured and saved for ulterior use (sometimes for up to 18 years or longer).
Calibrated: The devices used for monitoring purposes are showing the correct values. The devices are calibrated on a regular basis to ensure that the measurements are always showing the correct values.
The regulations don’t state how to fulfil regulations and as of such, the user can define how they qualify the areas, how they monitor the area and how often to calibrate the devices.
Working with the GxP guidelines
When a monitoring system is installed, the data generated must be evaluated to ensure that the temperature is always within the specific limits.
When using a stand-alone system, the data will not be live meaning that any temperature deviations that have occurred, will only be visible once the data is downloaded.
Within a continuous monitoring system, as soon as a limit is crossed, the user is alerted via E-Mail, SMS or telephone call and can act or react accordingly, knowing exactly where the alarm is, when it started, how long as it has been going on for and who has seen it.
The audit trail of the continuous monitoring system allows for users and auditors to track an area, seeing exactly what happened and when including all of the user information that has been added to the system, stating what standard operating procedures (SOP’s) have been carried out and also defining what corrective and preventive actions are required.
In any case, a temperature deviation means that something must be done!
Non-compliance
When an audit is carried out and the regulatory requirements not fulfilled, then a non-compliance is documented. The FDA Form 483 letter “Notice of Inspectional Observations” is a documented non-conformance after an FDA audit. Once a company has received a 483, the data is publicly available. In 2019, over 155 environmental monitoring issues (mainly temperature) were observed and documented in the fields of drugs, medical devices, human tissue for transplantation, veterinary medicine and biologics.

Controlled environments
When a facility is designed, an integral part of the design is the building management system (BMS). The BMS manages many services within a facility including the heating, ventilation and air conditioning (HVAC) of a building with the help of sensors located throughout the facilities. In order to ensure that the BMS is controlling the HVAC system correctly, an environmental monitoring system (EMS) can be installed. The EMS will monitor all the critical control parameters defined during the product risk assessment at critical locations defined during the facility qualification. 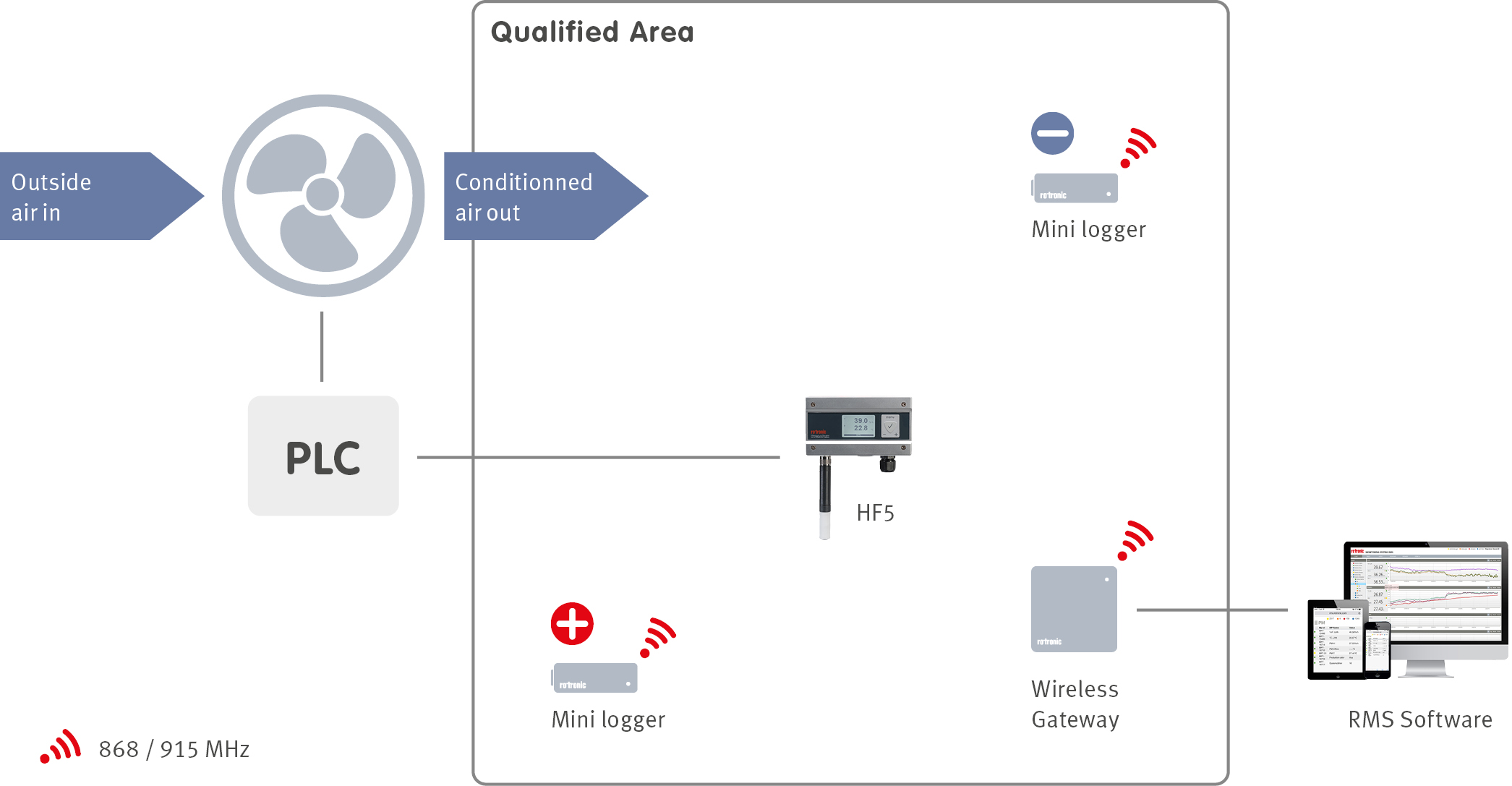
For controlled environments within the R&D phase as well as transportation and storage applications, the temperature will also be controlled. Monitoring is also required to ensure that the temperature control is being done correctly!
Qualify the area
The qualification of the area is to demonstrate that the area is fits its intended using scientific proof.
The USP 659 Packaging and storage requirements provides storage condition definitions relevant to the storage and distribution of active ingredients, excipients, and medical products, such as pharmaceuticals, devices, combination products and dietary supplements. All products should have specific directions in respect to the storage conditions (the temperature or humidity) at which an article must be stored and shipped (based upon stability studies).
The temperature and storage definitions for pharmaceutical products are the following:
- Freezer: -25…-10°C.
- Refrigerator: 2…8°C.
- Cold: 8°C
- Cool: 8…15°C.
- Room temperature: the temperature prevailing in a working environment.
- Controlled room temperature: 20…25°C. Mean kinetic temperature not exceeding 25°C, but excursions between 15…30°C are allowed, transient spikes up to 40°C are permitted as long as they do not exceed 24 hours. Spikes above 40°C may be permitted only if the manufacturer so instructs.
- What is mean kinetic temperature.
- Warm: 30…40°C.
- Excessive heat: >40°C.
- Dry place: average relative humidity >40% at 20°C.
- Protect from freezing.
- Protect from light.
WHAT IS MEAN KINETIC TEMPERATURE
The mean kinetic temperature is the total influence of temperature on an object or product over a certain period of time.
Extract from the ICH Topic Q 1 A (R2): Stabilty Testing of new Drug Substances and Products:
Mean kinetic temperature: a single derived temperature that, if maintained over a defined period of time, affords the same thermal challenge to a drug substance or drug product as would be experienced over a range of both higher and lower temperatures for an equivalent defined period. The mean kinetic temperature is higher than the arithmetic mean temperature and takes into account the Arrhenius equation.
Once the application of the area is defined, a temperature mapping is carried out to ensure that the entire room is controlled within the defined temperature range. This mapping will define hot and cold spots within the area that will then need to be monitored.
The WHO Supplement 8, temperature mapping of storage areas offers how to carry out a systematic mapping procedure in temperature-controlled areas.
THE USP 1079 also offers guidelines in terms of temperature mapping.
Rotronic offers devices for mappings, as well as rental kit and complete mapping services.
Install a monitoring system
The risk analysis will help define what kind of monitoring system is required and as of such the ISPE offer the GAMP5 Guide: Compliant GxP Computerized Systems to help regulated users as well as suppliers to deliver a suitable solution to meet the GxP regulations.
Rotronic offers the following electronic data loggers for monitoring purposes:
- HL-1D (stand-alone monitoring)
- HL-20D (stand-alone monitoring)
- RMS-MLOG (wireless continuous monitoring)
- RMS-LOG (wireless and wired continuous monitoring)
If the monitoring is done digitally, then the monitoring system should reply to the FDA 21 CFR Part 11 and EU Annex 11 regulations.
Rotronic offers the following GAMP5 category 4 software solutions that comply to the regulations for monitoring purposes:
- HW4 (stand-alone monitoring).
- RMS (continuous monitoring).
RMS Advantages at a glance
1) FDA 21 CFR Part 11/EU Annex 11 conform & designed based upon the GAMP5 guidelines.
2) Continuous monitoring.
3) E-Mail, SMS and telephone alarms.
4) Easy and intuitive to use.
The monitoring system (stand alone or continuous) will need to be validated ensure that it fulfils its intended use based upon the GAMP5 risk-based approach to compliant GxP computerized systems recommendations.
Choose a device
The USP 1118 Monitoring Devices – Time, temperature and humidity offers an overview of the measurement devices that can be used and explains the various technologies.
Rotronic employs the following technologies for the wired or wireless electronic data loggers:
Temperature sensors:
Relative humidity sensors:
Calibrate the device
The Rotronic devices can be calibrated using the device’s display or within the corresponding software. Rotronic offer a range of calibration devices for temperature and relative humidity:
- EAxx-SCS salt solutions (Delivered with an ISO-17025 calibration certificate)
- HygroCal100 humidity generator
- HG2-S humidity and temperature generator
- HG2-XL humidity and temperature generator
- S8000 dew point mirror reference device
RMS features a calibration management tool, where all historical calibrations are documented and reminders for upcoming calibrations are communicated. Rotronic also offer calibration services:
- On site calibrations.
- ISO 9001 calibrations.
- ISO 17025 calibrations.
Training
Rotronic offer hardware and software training to ensure that the users are fully capable of using our solutions for GxP applications.
Services
Rotronic is setup to help customers with the following services
Any Questions? Contact us today!
Recommended Products
-

WB-0001
The WB-0001 detects the presence of water or conductive fluids once it reaches a level that bridges the two conductive strips on the bottom of the housing.
Learn More -

RMS-MLOG-B-915 - Wireless Mini Data Logger - Temperature & Humidity
Small, inexpensive and flexible
Learn More -

RMS-MLOG-BT-915 - Wireless Mini Data Logger - Temperature & barometric pressure
Small, inexpensive and flexible
Learn More -

RMS-LOG-T30-868
The RMS-LOG-T30 is a data logger with two integrated analog-to-digital converters, to which two PT100 sensors can be connected for high-precision temperature measurement.
Learn More -

RMS-LOG-T30-915
The RMS-LOG-T30 is a data logger with two integrated analog-to-digital converters, to which two PT100 sensors can be connected for high-precision temperature measurement.
Learn More -

RMS-LOG-T30-L - Data Logger
The RMS-LOG-T30 is a data logger with two integrated analog-to-digital converters, to which two PT100 sensors can be connected for high-precision temperature measurement.
Learn More -

AC1207
For active adapter and converter cables Mains adapter RNG 11, 9 V / 200 mA, 3.5 mm stereo jack, tip + Learn More -

RMS T10-0113
The T10 temperature sensors are NTC (Negative Temperature Coefficient) thermistors, meaning that the resistance of the NTC decreases with increasing temperature. The T10 temperature sensors are compatible with the RMS-MLOG-T10-868/915 data loggers.
Learn More -

RMS T10-0013
The T10 temperature sensors are NTC (Negative Temperature Coefficient) thermistors, meaning that the resistance of the NTC decreases with increasing temperature. The T10 temperature sensors are compatible with the RMS-MLOG-T10-868/915 data loggers.
Learn More -

RMS T10-0006
The T10 temperature sensors are NTC (Negative Temperature Coefficient) thermistors, meaning that the resistance of the NTC decreases with increasing temperature. The T10 temperature sensors are compatible with the RMS-MLOG-T10-868/915 data loggers.
Learn More -

RMS T10-0005
The T10 temperature sensors are NTC (Negative Temperature Coefficient) thermistors, meaning that the resistance of the NTC decreases with increasing temperature.
Learn More -

RMS T10-0004
The T10 temperature sensors are NTC (Negative Temperature Coefficient) thermistors, meaning that the resistance of the NTC decreases with increasing temperature. The T10 temperature sensors are compatible with the RMS-MLOG-T10-868/915 data loggers.
Learn More -

RMS T10-0003
The T10 temperature sensors are NTC (Negative Temperature Coefficient) thermistors, meaning that the resistance of the NTC decreases with increasing temperature. The T10 temperature sensors are compatible with the RMS-MLOG-T10-868/915 data loggers.
Learn More -

RMS T10-0002
The T10 temperature sensors are NTC (Negative Temperature Coefficient) thermistors, meaning that the resistance of the NTC decreases with increasing temperature. The T10 temperature sensors are compatible with the RMS-MLOG-T10-868/915 data loggers.
Learn More -

RMS T10-0001
The T10 temperature sensors are NTC (Negative Temperature Coefficient) thermistors, meaning that the resistance of the NTC decreases with increasing temperature.
Learn More -

RMS-MLOG-T10-868 - Wireless Mini Data Logger - Temperature with external (NTC) probe
Small, inexpensive and flexible
Learn More -

RMS-MLOG-T10-915 - Wireless Mini Data Logger - Temperature with external (NTC) probe
Small, inexpensive and flexible
Learn More -

RMS-MLOG-T-868 - Wireless Mini Data Logger - Temperature
Small, inexpensive and flexible
Learn More